
Updated in October 2022
- Mnova MSChrom is a piece of desktop software for LC/GC-MS and tandem MS (or MS-MS) processing, analysis, and reporting.
- One software fits all:
- Opens the raw datatypes used by various LC/GC-MS vendors
- Can work together with NMR and IR/UV data seamlessly
- Visualization, annotation, and reporting made easy
- PowerPoint-like, multi-page, and multi-document graphical interface
- Setup of initial display: TIC, mass spec, UV traces, and alignment of traces
- User-defined display templates for easy reporting
- Powerful analysis tools for:
- Extracted ion chromatograms to locate expected ions
- Automatic peak integration with easy manual correction
- Elemental composition enumeration, molecule match, mol. ion and isotope cluster prediction
- Molecular match that automatically matches the expected molecular ion and isotope cluster patterns starting from a molecular structure or MF with different adducts and losses considered
- Elemental composition enumeration from high-resolution mass spectrum
- Isotope cluster prediction starting from a molecular formula, with different adducts/losses considered
- Batch or real-time data processing and databasing
- Scripting engine included (javascript)
- Real-time or batch processing and reporting (with Mnova Gears)
- Database management of raw data and analysis results (with Mnova DB)
- Communication with ELN (Customization development with Mestrelab)
Learn how to get your first results in a few minutes with Mnova MSChrom:
The scope of this guide is to get you started in the use of MSChrom. For further information please check out our manual that you can find on either of our downloads and Mnova pages.
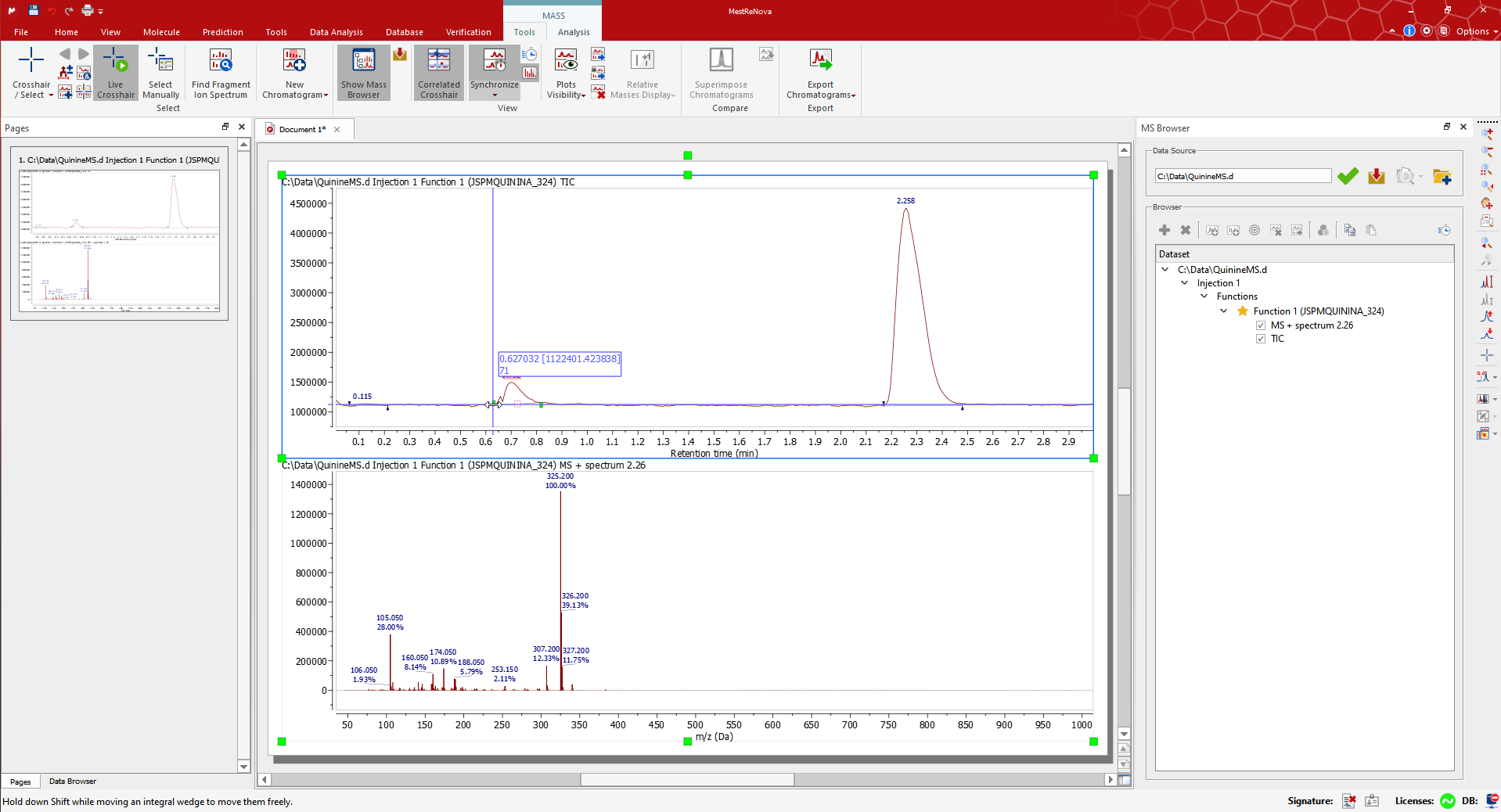
Drag and drop the raw file to Mnova to display the TIC and MS spectrum of the highest signal. Alternatively use ‘File > Open’). The MS Browser will be automatically displayed, and the Mass/Tools ribbon automatically activated. For example, open the QuinineMS.d dataset from the Mnova Examples*.
*Note: You can find the Mnova dataset examples included in the application by following the path 'Help > Download Examples'

If it is available, the PDA total absorbance chromatogram will be displayed at the top of the page. (TIP: The data to display when loading a new mass item can be configured in the ‘Setup’ tab for the Mass Preferences).
The MS Browser mentioned above contains very useful information, for instance about your LC-MS experiments. You can open the TIC, Base Peak or the Mass Spectrum through the Function section, extract an UV trace by clicking on the DAD Trace, and generally navigate through all your data.


Use the 'Zoom in' tool to show details in any plot. (Use the 'Z' Key as a shortcut or the corresponding 'View' icon on the toolbar).

Manually edit the peak range by dragging the integral wedges that indicate the peak range (hold down 'Shift' while moving a wedge to move them freely).
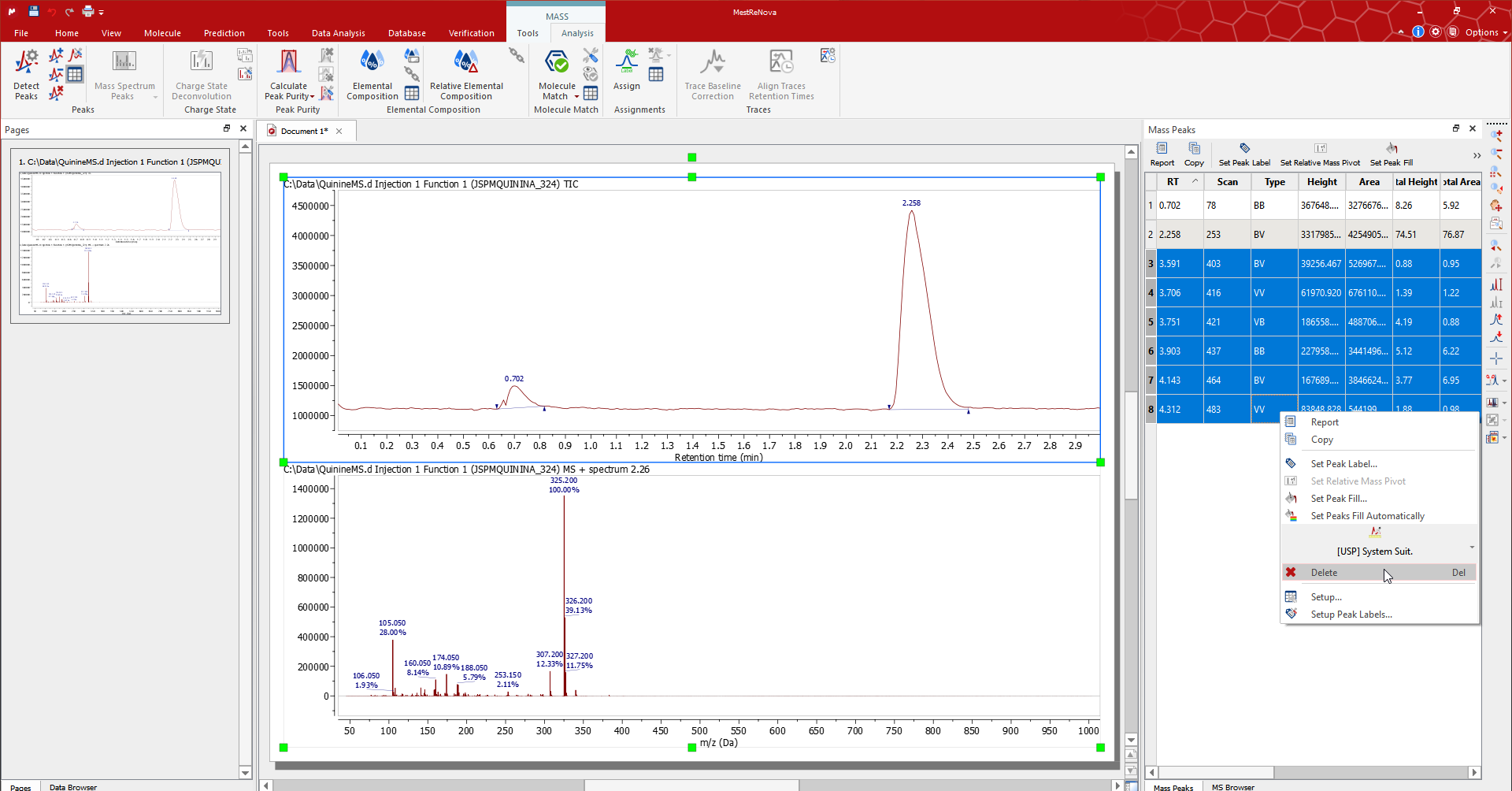
The peaks that Mnova automatically detects can be edited using the tools for the Peak group in the 'Mass/Analysis' ribbon or in the peak context menu (right click on the peak label).

The Mass Peaks table contains the details of the list of peaks for each plot. Edit/remove the peaks by selecting the associated rows in the table.
Select another mass spectrum at a particular retention time value or range with the 'Crosshair/Select' feature in the Mass/Tools ribbon.

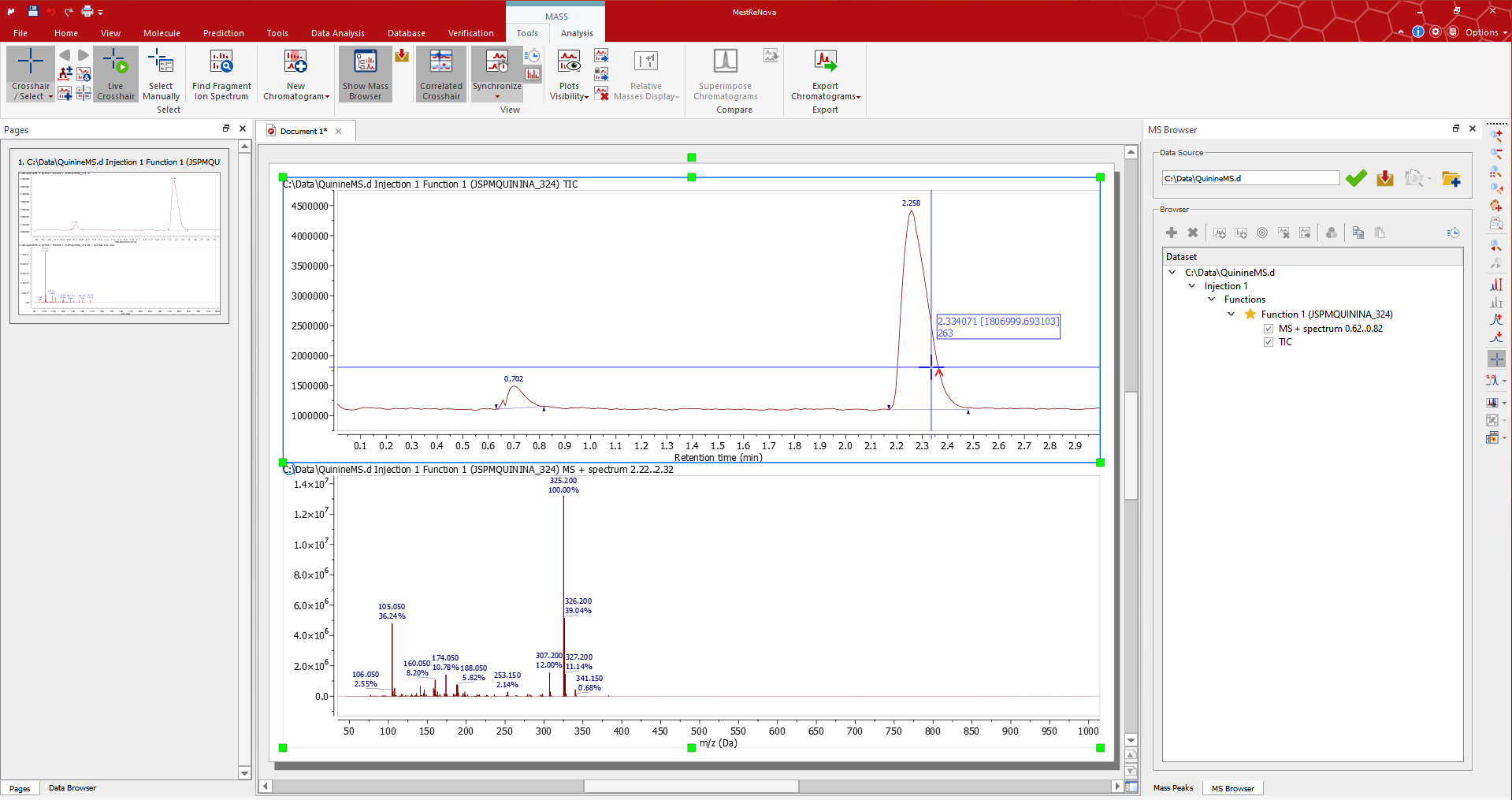
Switch to Crosshair/Select Manual: A preview of the MS is displayed when hovering the mouse over the TIC if 'Live Crosshair' is active. Click on the TIC to display the mass spectrum at the desired retention time. In this example, we have manually selected the spectrum at a retention time of 0.70 min.

If you need to average the mass spectra over a certain time range (co-add), switch to Crosshair/Select Manual mode and click-and-drag on the TIC to display co-added spectra in the desired response time range. In this case, we co-add multiple mass spectra from 0.64 to 0.82 (the label at the top of the MS will inform you about the range of retention time).

The Crosshair/Select Peak mode allows you to co-add a whole peak. Click on a peak to display the co-added MS within the peak range.
The Crosshair/Select Peak (Background subtraction) mode displays the co-added MS within the peak range with the background subtracted.

Toggle on Appending mode to generate a new mass spectrum when using the Crosshair/Select tool.

For simplicity, let's undo all these changes and load the QuinineMS.d dataset again.
Display the Extracted Ion Chromatogram (EIC) for an MS peak using the New Chromatogram tool from the Mass/Tools ribbon. The cursor will be displayed in the mass spectrum plots. Select an m/z value or click-and-drag for a range. The corresponding EIC plot will be appended at the bottom.
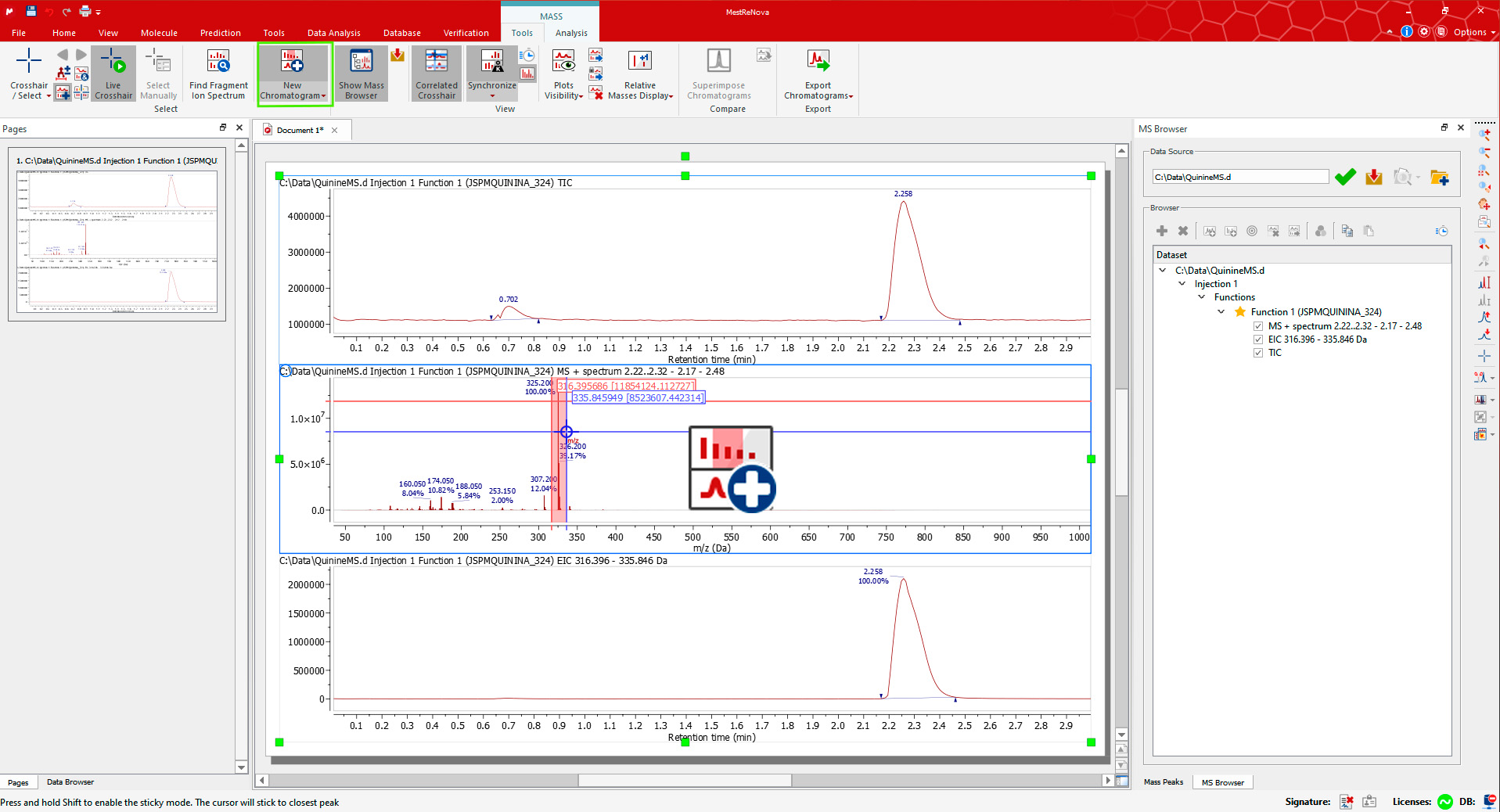
Let's now reload the QuinineMS.d dataset.
The Molecule match analysis determines the possible presence of one or more given structures (or molecular formulae) within a mass spectral dataset. The isotope cluster for each structure is computed and compared to each spectrum in the dataset, and the mass spectrum with the best match that returns a score above the preset threshold is taken as a positive “match” or will otherwise be taken as "Not found".
Open one or more structures in the current document. Click on the ‘Molecule Match’ button of the Mass/Analysis Calculate Molecule Match, and the Molecule Match table will be automatically displayed. This table contains the status of each structure or formula “matched” or “not found” - and other information such as Retention Time, Match Score, Adduct/Lose, etc.

By clicking on any matched row in the table, the Matched Isotope Cluster Chromatogram for the corresponding molecule will be overlaid as a blue line in the TIC: it indicates where the best match is. The mass spectrum corresponding to that retention time (2.26 min in this case) is also displayed with the predicted isotope cluster overlaid on it (the blue shaded area plus blue peaks with 'whiskers').
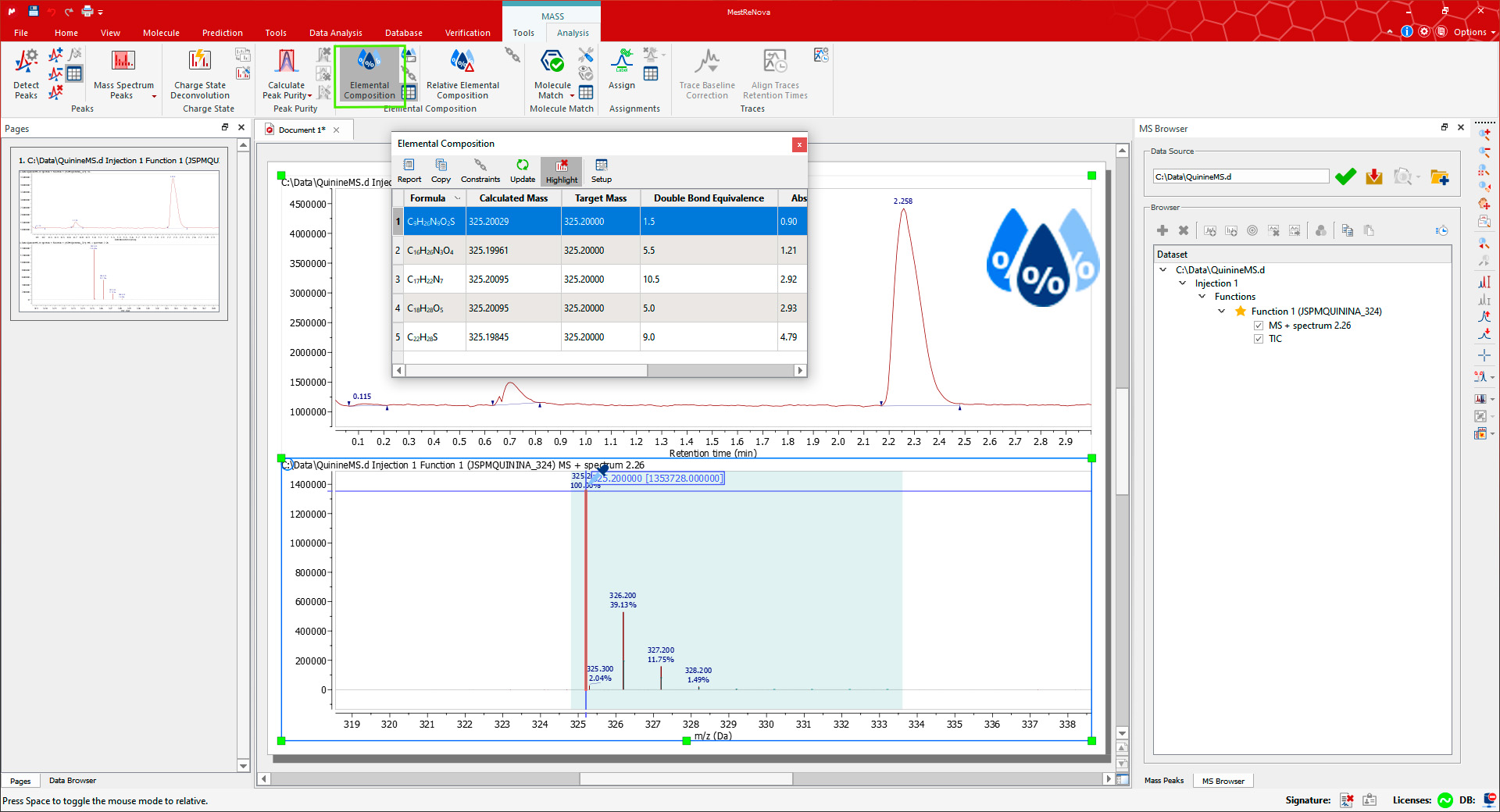
If you are using a high-resolution MS, it is possible to do an 'Elemental Composition' analysis of any peak in the mass spectrum. First, open the Elemental Composition Constraints dialog using the Constraints button on the Elemental Composition group in the Mass/Analysis toolbar.

You can always add more 'Element Constraints' just by clicking on the highlighted 'Add' button. Once you have finished with the constraints, click on OK and go back to the MS spectrum to click on the desired MS peak with the Elemental Composition selection tool (which activates the cursor). The Elemental Composition table is automatically shown with the results on the selected mass peak.
You can predict the isotope cluster of a molecular formula with different adducts and losses using the highlighted tool 'Predict' located in the MS Prediction toolbar. Add or choose a molecular formula and click OK to simulate the isotope clusters with the predefined adducts/losses.

The Mass Prediction table is shown. Click on the row with the desired Adduct or Loss and the prediction will be overlaid on the active mass spectrum to allow its comparison with the displayed mass spectrum.

Finally, you will be able to save, print, or export the document to PDF by clicking on the corresponding icon in the File menu.

You can check the following pages for MSChrom plugin features and resources and details about supported data formats.
And once again, if you wish to read more information about how to use MSChrom in more detail please check out our manual that you can find on either of our downloads and Mnova pages.
