
Mnova 8.0 is a release with very significant new functionality, plugins and important changes to many functions in the program.
- Designed to deliver greatly improved results.
- The new Mnova Verify module for Automatic Structure Verification Improvements to the NMR plugin, including assignments, peaks and multiplet analysis.
- Improvements to MS plugin which can now extract UV spectra.
- New Molecule Editor
-
New Features
Enhancements in the Assignment Module.
1. Auto-assignments Mnova provides a very simple interface to automatically assign your molecules to the applicable 1H-NMR spectrum. Mnova will automatically run a multiplet analysis and will fully assign your spectrum to the corresponding molecular structure *Please note that this feature needs a license of the Mnova NMR and Modgraph NMRPRedict Desktop plugins

2. Feature to swap assignment It will be possible to swap assignments if previously they were wrongly assigned as illustrated in the figure below, where assignments (3) and (6) must be swapped.
 3. Capability to assign to several compounds It will be possible to assign the same spectrum to different compounds in a mixture and to select a different color for the assignments of each compound. In the example below the assignments labels of the quinine appears in blue and the assignments labels of the EtOAc are showed in green:
3. Capability to assign to several compounds It will be possible to assign the same spectrum to different compounds in a mixture and to select a different color for the assignments of each compound. In the example below the assignments labels of the quinine appears in blue and the assignments labels of the EtOAc are showed in green: 
4. Capability to change atom labels and numbering from the Assignment Table It will be possible to change the label of a -CH2 group, just by double clicking on the applicable ‘atom cell’.
 You can also change the atom number by double clicking on the applicable atom cell. 5. Export Assignments as ASCII It will be possible to export the assignments as an ASCII file, just by following the menu ‘File/Save As/Script: Assignments’
You can also change the atom number by double clicking on the applicable atom cell. 5. Export Assignments as ASCII It will be possible to export the assignments as an ASCII file, just by following the menu ‘File/Save As/Script: Assignments’Enhancements in the Peaks
1- Capability to select the compound for each peak You will be able to select the compound for each peak from the Peaks Table or by right clicking on the Peak label and selecting ‘Set Peak Compound’
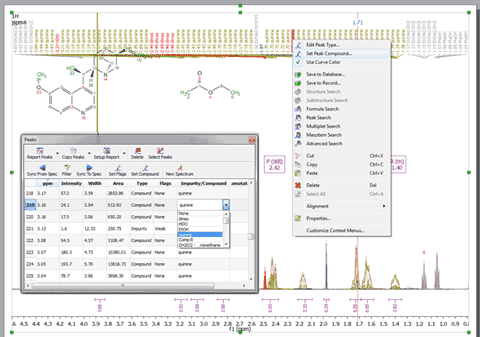
2- Capability to show Peaks Table only for a given compound If the spectrum contains peaks belonging to more than one compound, we can show only the peaks assigned to one of them (by using the ‘Filter’ button):

3- Capability to create a synthetic spectrum from the Peaks table The ‘New spectrum’ button will generate a synthetic spectrum from the selected peaks in the table or for all the peaks shown in the table if there is no selection.

4- Capability to merge peaks It will be possible to merge peaks (very useful for 2D) just by selecting the applicable option in the Peak Picking scroll down menu and clicking and dragging on your spectrum. 5- Capability to edit Peak Type at the multiplet level Right clicking on the multiplet box will allow you to change the ‘Peak Type’ (from compound to impurity, solvent, artifact, etc) or to assign the peaks inside the multiplet to a specific compound

6- Fit to the highest compound This new feature will adjust the display of spectral intensity in both 1D and 2D spectra so that the strongest compound signal is optimally displayed (discarding solvent or impurity peaks) 7 -Improvement in the Absolute Referencing Dialog box. All spectra are now automatically checked
Enhancements in the Multiplet Analysis Feature
1- Capability to export multiplets into spin simulation module The table of multiplets can be used for the simulation of a synthetic spectrum.

2- Converter to simulate spectra from lists of multiplets in XML format. It will be also possible to synthesize a spectrum, just by loading a XML file with information about the multiplets. 3- Display multiplet boxes for a specific compound only If the spectrum contains peaks belonging to more than one compound, we can show only the peaks assigned to one of them by filtering the desired peaks in the Peaks Table. Checking the ‘Follow Peak Visibility rules’ box will allow you to show only the multiplet boxes applicable to those visible peaks.

4- Display the Multiplets Integrals by default The multiplet integrals curves are shown by default in Mnova 8
2D and 1D Spectra interaction
1- Capability to drag&drop 1D spectra over a 2D dataset to use them as external traces It will be possible to drag and drop any currently opened 1D-NMR spectrum to a 2D NMR dataset in order to get them as traces. Here you can see how the page number 1, containing a 1H dataset is dropped to the HSQC to get the horizontal trace:

The same can be done with the page 2 (containing a 13C spectrum) to get the vertical trace:

2- Automatic Referencing of 2D spectra with 1H external traces It is possible to automatically reference a 2D-NMR spectrum directly with the 1H external trace, from the ‘Setup Traces’ dialog box:

Other New Features
1- New Whitening phase correction algorithm for 1D NMR datasets Mnova incorporates a new method for fully automated phasing of Fourier transform NMR spectra based on a combination of the whitening and the metabonomics method.
 2- Ability to customize default phase correction method You can select by default an automatic phase correction made by Mnova or to use the phase correction imported from the NMR spectrometer.
2- Ability to customize default phase correction method You can select by default an automatic phase correction made by Mnova or to use the phase correction imported from the NMR spectrometer.
3- Spectrum resolution added to ‘Info View’ window of Crosshair The Crosshair window will show the spectrum resolution:

4- Add class column in the Stacked Spectra and Parameters table In the context of metabolomics, you would need to classify the stacked spectra by specifying a class in the ‘Stacked Spectra Table’.

5- Capability to select/deselect available series on a graph plot. Right clicking on the Data Analysis Graphic, will allow you to update the graph after having made changes in the Stacked Spectra Table or directly on the stacked spectra (very useful, for example if you invert the order of the spectra). If you have more than one curve on the Data Analysis Graphic, you will be able to select/deselect available “series” (curves) on the plot.

6- Import XY data (txt and csv format) into Data Analysis It will be possible to fill the Data Analysis panel just by loading the XY data in txt or csv format 7- Capability to guess the kinetic reaction order Mnova will try to guess the order of the reaction, fitting the data to standard equations and comparing the mean square error.

8- ‘Average Sum’ mode for the binning The ‘Average Sum’ will divide the sum by the number of points on the bin

9- Capability to show the baseline from the Properties dialog box It will be possible to draw the baseline by selecting the applicable option in the ‘Properties/Grid’ dialog box (very useful if you have removed the vertical scale):

10- Ability to use imported or automatic Apodization values From the preferences menu, the user will have the capability to import the apodization and baseline correction made on the spectrometer

More New Features
- Script to obtain directly the FID (without any processing)
- Capability to save Blind Regions to a recent saved name
- Capability to delete saved Blind Regions
- Alignment regions can be loaded/saved as xml from Align Spectra dialog
- New converter for Prospa data files
- New NMRPipe Converter for 2D-NMR datasets
- Improved the speed to read files over network
- Special script converter to export spectra from arrayed item as an ASCII matrix
Bugs Fixed
- Disabled points in the data analysis graph were copied to the clipboard
- Incorrect labels when the assignment was changed
- Problems correcting the bias and slope from the integral manager
- Auto tuning did not work right if the tuning range was too big
- The multiplet labels were changed incorrectly when the user edited the atom number of an assigned atom.
- Full View did not update when browsing to next multiplet from Multiplet Manager
- Assignments introduced by typing in the assignments table could not be copy&pasted
- Problems disabling Blind regions
- Problems normalizing a spectrum to a negative area
- Problems saving dipolar and quadrupolar constants in spin simulation mode
- Problems simulating spectra with too many Js
- gHMBCAD from Varian Chempack was not process correctly
- Autophase correction was applied before blind regions
- Multiplet label not updated when reassigning a hydrogen
- Report Special moved to the right side of main window
- Manual zoom fails in 2D stacked mode with different SW in F1
- Manually deleted peaks got back after going to peak picking options
- Values could not be continuously changed by keep pressing the arrow buttons in Line Fitting table
- Wrong units for big and small delta of a DOSY in JEOL format
- Multiplet integral not re-calculated when adding or deleting peaks to/from it
- Wrong peak labels when copying&pasting a spectrum
- Problems opening old WinNMR files
- Option to display Peak Annotations was not persisted in the Default Settings
- Problems saving a formula in Arithmetic tool
- Using existing integrals when doing auto multiplet analysis
- PP threshold doesn’t refresh when changing PP Noise Factor
- Not all 2D assignments were displayed if they were overlapped
- GSD curves dissapeared when saving/loading a document
- Multiple selection in Line Fitting was broken
- 2D Peak Picking did not flag solvent peaks as Solvent
- Wrong Lowest Frequency for a JCAMP dataset
- Wrong processing of HSQC spectra with ‘f1coef’ parameter empty
- Multiplet integral labels displayed vertically
- Manual phase dialog: could not overwrite the PH0 and PH1 values easily
- Restored cuts appeared again in stacked plots
- Use Current button disabled in Integration Options of the Processing template
- Layout templates didn’t work if page navigator was hidden
- Magnitude in f1 was lost after phasing f2
- Problems with Absolute Referencing Feature when the HSQC was in the first page
-
New Features
- New J-predictor engine for substituted aromatic compounds
- Calculate J-coupling values not calculated by Modgraph
- Capability to select specific prediction Databases
- New method to calculate Modgraph Predictor desktop error interval
Bugs Fixed
- Problems highlighting atom(s) when cursor was on predicted peak
- Error when trying to remove a molecule from the 1H DBA
- Error when trying to update C13 prediction DB
- Problems highlighting peaks of a predicted NMR spectrum
- Error message when saving a change in 13C Prediction DB
- Wrong labels in the highlighting of the equivalent atoms
- Predicted DEPT including not protonated carbons show signals in a wrong side
- 15N-prediction didn’t work for molecules with charges
- Error predicting 15N NMR of a molecule
- Predict and Highlight showed highlighting at 0 ppm for all nuclei
- Wrong assignments shown in the peak labels
- No highlight in Compounds table when hovering the mouse over a peak of a predicted spectrum
- Problems predicting the NMR of allenes with a carbon drawn as a dot
- Wrong prediction when mol file has two molecular blocks
- Wrong 31P and 19F predictions of a molecule
- GSD peak picking method used when running Predict and Verify
-
New Features
Capability to extract UV spectra by Alt+Clicking over a peak in a DAD chromatogram
Using the crosshair mode and clicking on a UV chromatogram while maintaining Alt pressed, will display the UV spectrum for the clicked retention time.

Calibration of chromatograms with external traces
For some technical reasons, the TIC peaks can be delayed compared to the UV peaks. This feature will re-calibrate the chromatograms so that the peaks match across them. You will only need to click on a peak in the selected chromatogram and enter the new value to change the X scale (time).

UV subtraction
Similarly to the existing chromatogram subtraction, there is a new script for UV subtraction which will subtract a background UV chromatogram from a foreground one.

Bugs Fixed
- No peak labels in MS spectra after running MolMatch
- Problems displaying some ES- functions
- Lanthanides and Actinides missing in the Elemental Composition constraints
- Problems running a Massitem Search without MS spectrum and with floating licenses
- Minimum Area Threshold in Peak detection options could not be higher than 1
- Wrong peak integration in UV chromatogram
- Report of MolMatch and Molecules tables got pasted several times
- Toggle Centroid Spectrum did not work with full MS dataset saved documents
-
New Features
Search for peaks with a specific compound label
If your spectrum has more than one compound, you can run peak searches of only one of the compounds:

Save All Compounds with their Peaks to different records in the DB
If your document contains more than one compound and several molecules are selected for saving, a warning message will be displayed:
 When the user chooses to continue, several records (one for each selected molecule) are created in the database and each of the records will contain all of the selected spectra.
When the user chooses to continue, several records (one for each selected molecule) are created in the database and each of the records will contain all of the selected spectra.Other New Features
Molecule fields (Molecule name, label, list of aliases) are accessible by scripting Improvement to the Database Preview dialog Objects saved in Mnova DB are associated with its page number
Bugs Fixed
- A new button to ‘Rerun Query’ has been added in the Hits dialog and in DB Browser which allows you to refine your search.
- A new button to ‘Convert to Advanced Query’ has been added in the Query Editor dialog. In case you start with a simple search (not advanced), it is possible to convert it to an advanced search so that you can add new queries and combine them.
- Problems showing the NMR Preview of a second NMR spectrum in DB Browser
- MnServer closed connection on reading molfile with very large or very small xyz values
- Objects saved in Mnova DB were not associated with its page number
- Problems saving twice a big stacked spectra
- Hits excluded because of exceeding the maximum hits are not used in an advanced search
