
-
New Features
Capability to process Varian DEPT spectra.
Mnova is now able to open the new Varian DEPT spectra containing two arrayed variables “mult” and “qphase” (with 8 slices).

Improvements in the assignments module
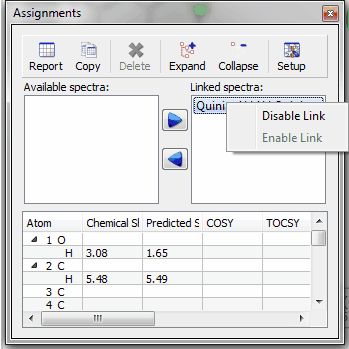
Capability to make assignments to a group of atoms Several atoms in a molecular structure can be selected at a time by holding down Ctrl key and clicking.
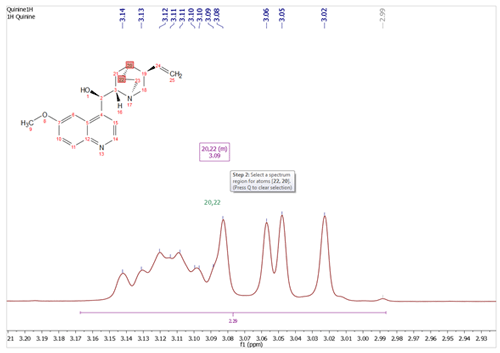
Added the ability to hide/show and delete all the assignments of a linked item or molecule It will be possible to hide and delete the assignments in a spectrum, just by right clicking on the molecule and selecting the applicable option.

You will also be able to hide the assignments by right clicking on the ‘Linked Spectrum’ from the Assignments table and selecting ‘Disable Link’.
Capability to assign the 2D correlations keeping the original chemical shifts from one of the 1D If you are assigning a 2D dataset and you had already assigned the atoms in the 1D datasets, you will obtain a dialog box like the one below, which will allow you to keep the assignments from the 1D, replace or add new assignments.

Assign Dialog keeps the option that user clicked last time for each dimension The latest ambiguous assignment option selected will be stored in the registry.
Improvements in the Multipoint Baseline Correction
The dialog box has been simplified, removing the Options button and showing the Algorithm used.

The settings used last time are kept in the dialog. Hovering the mouse over a baseline point, will show a tooltip which reads: “Click and drag to move the point” and “Double click to remove the point”.

Other new Features
Ability to set up a minimum width for the stacked scale. It will be possible to select the margin of the stacked scale from the Properties dialog box:

Line widths for 1D display and 2D contours independent. From the Properties dialog box, it will be possible to select different ‘Line Width’ values for 1D and 2D datasets.
Limit the number of digits shown in the X(I) column of the Data analysis panel. We have reduced the number of significant digits to avoid overcrowded cells in the Data Analysis panel.
Normalization of spectra excluding cut areas. Cut regions of the spectra will not be taken into account in the normalizations.
Include new peaks inside a multiplet when expanding the multiplet range. The new generated multiplet obtained after having changed its range, will include all the peaks inside the new range.
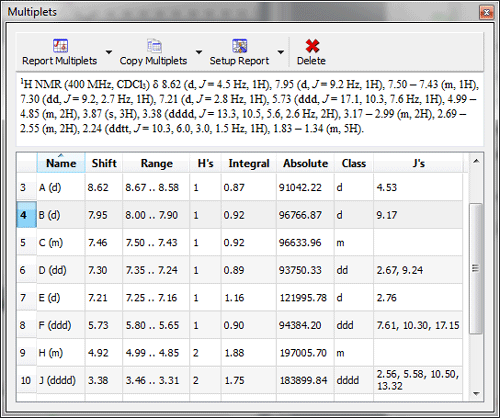
Multiplet Manager displays the absolute integral value. Information about the absolute integral value has been added to the Multiplet Manager dialogbox; which will be very useful for qNMR, to calculate the concentration conversion factor.

Multiplets Table displays absolute integral values of each multiplet A new column for the Absolute integral values has been added to the Multiplets table. By default, this column is hidden and it will be placed after the Integral column when it is visible.
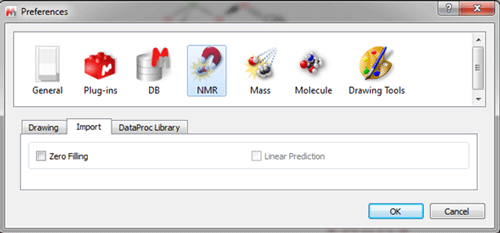
Options to import ZF and LP are disabled by default in the Preference menu.
Bugs Fixed
- Highlights of assignments do not update numbers in case of changed atom numbers
- Atom in molecules table is not highlighted when hovering the mouse over assignment label in the spectrum
- Color scheme of stacked 2D spectra changes when hiding some of them
- Free Selection did not work in the Multipoint Baseline Correction panel
- title did not retain the original color even if the spectrum was hidden
- Problems creating a Data Analysis Graph after deleting some spectra in the arrayed spectra
- J-tree labels too big after having exported to pdf with high resolution
- Capability to move the multiplet segments up and down
- Edit Blind Region Rule displays the blind region immediately
- The Undo command after changing the color of one spectrum in overlaid mode did not restore the original colors
- Wrong exporting of assignments to SDFile
- Assignment visualization problem in HSQC spectrum
- Antialiasing not turn off when copying as image
- Color scheme gets modified when changing the order of a stacked spectra
- Link information was lost when stacking spectra
- Fix the canvas item wrapper to give access to type and subtype properties of a layout template item from scripting
- Type of 2D traces could not be selected in Properties dialog
- Assignment labels not displayed when the scale was not in ppm
- Wrong list of HSQC correlations for methyl goups
- The tables of assignments and predictions are sorted by atomNumber instead of atomIndexes
- Correlations not pasted when copying and pasting NMR Assignments
- Problems reporting assignments from a DQF-COSY
-
New Features
Capability to select the algorithm (best/charge/increments) for shifts and Js.
It will be possible to select different algorithms for the prediction of chemical shifts and coupling constants

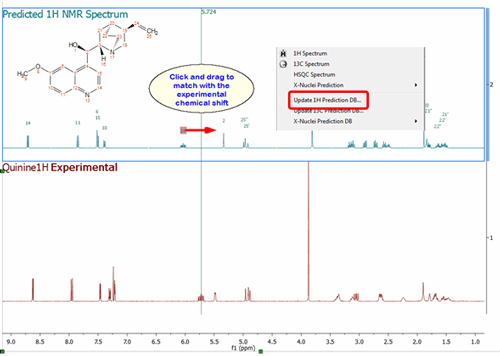
Updating the Prediction Database with the assignments of the predicted spectra.
It will be possible to update the Prediction Database directly from predicted spectra, which is very useful in those cases where the user has modified the prediction to fit the results with the experimental dataset in the Predict&Compare mode:
Different titles for predicted and simulated spectra.
The synthesized spectra generated with the Spin Simulation feature will be titled as ‘Simulated NMR Spectrum’
Bugs Fixed
- HPrediction labels from simulated spectra not shown when pasted as metafile
- 1H Prediction Table: Incorrect highlighting in the Ungroup view.
- Problems highlighting crowded regions in 1D and 2D predicted spectra
-
New Features
Capability to select multiple chromatograms and zoom all together.
It will be possible to select multiple chromatograms of the same dataset to zoom in/out simultaneously. The chromatograms can be selected as in a stack of NMR spectra:
- To select a single spectrum, left click on the spectrum while keeping pressed the Alt key
- To deselect a spectrum: left click + Ctrl + Alt
- In order to select multiple spectra, follow the same operation as above while keeping pressed Ctrl and/or Shift keys.

-
New Features
New Scripts to read annotations info and to create Databases from SDF files

Script to read annotations info from SDF files. SDF files may have several columns with annotation information (such as authors, project names, compound ID etc). This new script will read that information.
Create database definition file from SDFile This script will input a SDFile and output a database definition .xml file. The Database definition .xml file can then be used to create new databases. The SDFile can be imported using Scripts/Database/Import SDFile, preserving ALL tags and data.
Improve mapping of Mnova document items to db items. It will be possible to store and retrieve anything to/from the Databases. For example, now it will be possible to storage/retrieve ‘page notes’ or ‘individual mass item plots’ which was not possible in previous versions. Advanced Search now appears in the context menu The Advanced Search feature has been added by default to the context menu

Other new features
View hits against spectrum which originated query It will be possible to show the original spectrum (or molecule) used for the search to be compared with the hit from the Database. In the picture below, you can see the original spectrum used for the search to the left and the hit from the Database to the right

Script to auto MS search for all TIC peaks in the Database The Search MS in Chromatogram script, will search for all the mass spectra under each TIC peak. You will only need to load your GC/MS dataset and after having run the script, a dialog box will be displayed asking for the options for matching MS peaks:
The script will get the mass spec under each TIC peak, do peak picking, and search for the peaks against the selected DB.
Bugs Fixed
- Wrong number of items and records reported by Import Pages script
- Error when saving or retrieving big spectra in a Mnova DB
- Cutting on MS spectrum did not change the peak MS query
- Data integrity issues with binary object data
- No dialog should show when storing a document with duplications of the same molecule
What’s new in Mnova 7.1.1
0
Share.
